Becton Dickinson IV System Recall Issued After Patient Fatality
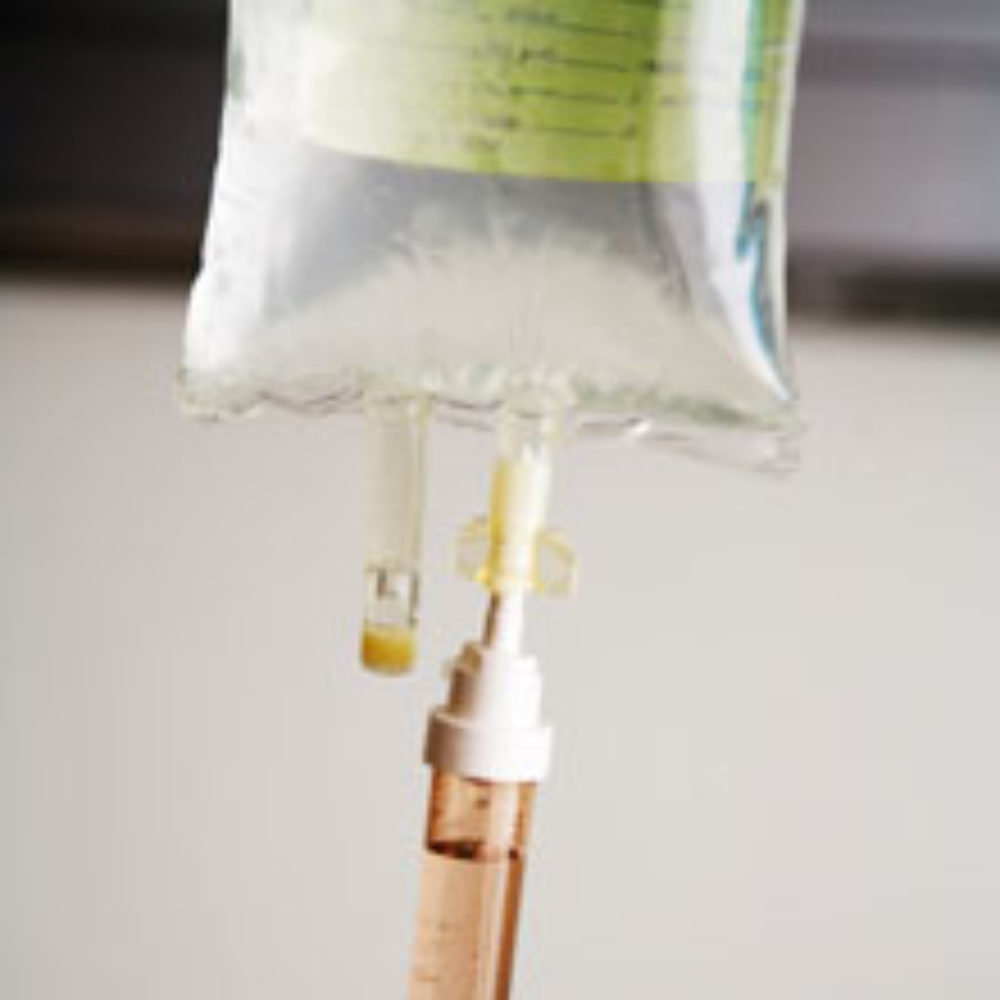
A medical device company is recalling 7.8 million catheters and intravenous (IV) fluid delivery system components after receiving reports of at least one possible death related to the products.
Becton, Dickinson & Co. (BD) announced that it is recalling certain lots of its BD Q-Syte Luer Access Devices and BD Nexiva Closed IV Catheter Systems because of a defect that could cause a patient to suffer an air embolism. Air embolisms, which occur when a bubble of air gets into the blood and travels through the heart, can result in serious injury or death. The devices also have the risk of blood leakage or the leakage of medication that should be delivered into the body, which can also lead to injuries and death.
While BD’s announcement made no mention of actual injuries or deaths reported, the Wall Street Journal reports that the recall was issued after the company received reports of a patient death last Thursday. The death, and another reported serious injury connected to the devices, have not been confirmed as being caused by the recalled IV components.

Did You Know?
Millions of Philips CPAP Machines Recalled
Philips DreamStation, CPAP and BiPAP machines sold in recent years may pose a risk of cancer, lung damage and other injuries.
Learn MoreThe components link needle-less IV catheters and medicine pumps to syringes, IV administration sets and other devices that introduce medication or draw blood from the patient’s body. The recall involves 2.8 million Q-Syte devices, and 2.9 million Nexiva units. The Nexiva units have two Q-syte devices packaged with them that could be affected by the recall.
The announcement is an expansion of a previous BD catheter recall, issued on October 28, 2009. The company says that the problem was caused by a manufacturer’s deviation that causes air to enter through the bottom disk of the septum of the devices. A list of catalog and lot numbers of devices affected by the recall expansion is available in BD’s press release. All of the lots affected products were distributed from November 2008 through November 2009.
The company says that it has notified customers worldwide by letter. Anyone experiencing adverse effects from the use of these devices should contact the FDA’s MedWatch Adverse Event Reporting program at www.fda.gov/medwatch/report.htm.
Get more articles like this sent directly to your inbox.
"*" indicates required fields


0 Comments