Stryker Receives FDA Warning Letter Over Manufacturing Process

Following an inspection at one of its production facilities, the FDA issued a warning letter to Stryker Corporation over quality issues involving its medical device manufacturing practices.
Stryker disclosed that it has received the warning letter in a press release issued today.
The company indicates that the FDA issued the warning after it inspected the company’s Portage, Michigan facility.

Learn More About
Lawsuits are being reviewed for several different dangerous and defective hip replacement systems.
Learn More About this Lawsuit See If You Qualify For CompensationThe letter cites the company for failing to notify the FDA of a medical device recall, but does not name the device that was recalled. The company also says the FDA criticized the company’s marketing of the Neptune Waste Management System, which it did without receiving a 510(k) approval.
The 510(k) fast track approval process is one of the least stringent means of gaining approval for a medical device, requiring only that the company show that the device is substantially equivalent to a medical device already on the market.
The company says it is fully cooperating with the FDA and has submitted corrective action plans to address FDA concerns.
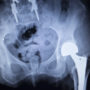
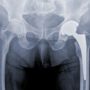
Stryker currently faces a growing number of lawsuits over its Rejuvenate hip replacement system, which was recalled from the market last year amid reports that a higher-than-expected number were failing within a few years.
All of the Stryker Rejuvenate hip lawsuits involve similar allegations that plaintiffs experienced problems where the modular hip neck-stem was corroding, fretting and failing as the metal parts rub against each other during normal, daily activities.
Get more articles like this sent directly to your inbox.
"*" indicates required fields



0 Comments