FDA Hopes To Improve Surgical Staplers Safety Through New Rules And Guidance
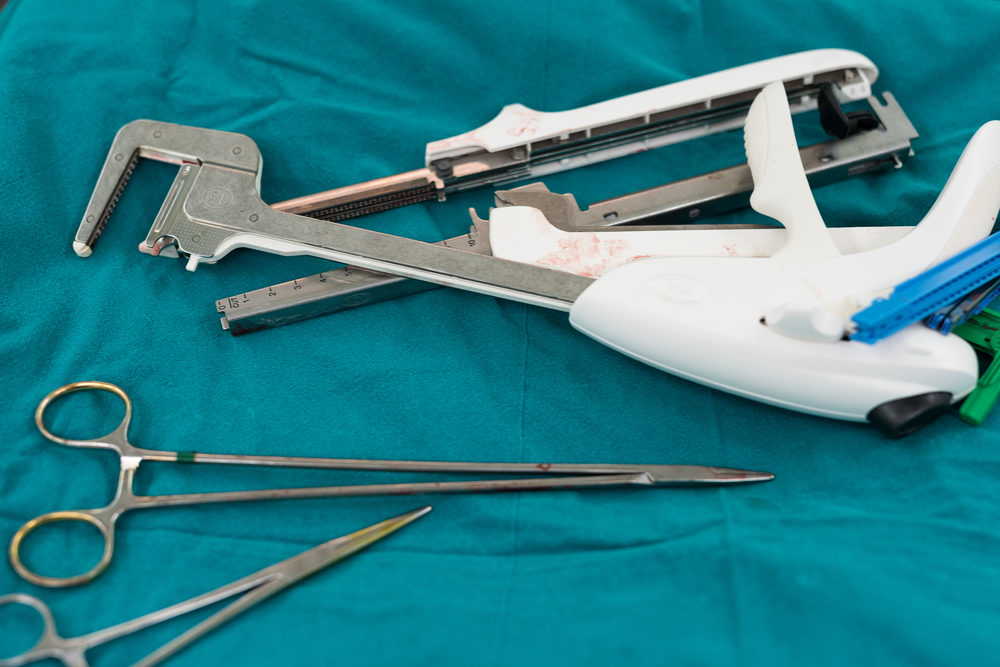
To address growing concerns, federal health regulators want manufacturers to put more stringent label warnings on surgical staplers, and to classify them as moderate risk medical devices, increasing the regulatory requirements for getting new ones approved for sale.
On April 24, the FDA published a proposed rule to change the classification of surgical staplers from low risk to moderate risk medical devices in the Federal Register. The agency also published a draft guidance that urges medical device manufacturers to be more thorough in explaining the potential risks, complications and contraindications linked to surgical stapler use.
The two actions come following an FDA warning to doctors in early March about the high numbers of surgical stapler complications and patient injuries reported in recent years. According to the agency, it received more than 41,000 reports of adverse events between January 2011 and the present.
Learn More About
Problems with surgical staplers may result in devastating injuries from internal surgery staples. Lawyers review cases nationwide.
Learn More About this Lawsuit SEE IF YOU QUALIFY FOR COMPENSATIONThe most commonly reported problems linked to surgical staplers involve an opening of the staple line, malformation of staples, staplers misfiring, staplers being too difficult to fire, staplers failing to fire and staplers that were misapplied. The FDA warning indicates that stapler malfunctions or misuse can cause prolonged surgical procedures, the need for additional surgeries, and other complications; such as bleeding, sepsis, internal organ damage, and death.
“Alerting the public and health care professionals was an important step, but as we shared at that time, we believe additional actions must be taken to better ensure the safe and effective use of these devices,” Dr. Jeffrey Shuren, director of the FDA’s Center for Devices and Radiological Health, said in an April 23 press release. “To that end, today we are announcing three new efforts with the goal of better protecting patients from the malfunctions, injuries and deaths associated with these devices.”
The proposed rule changes the classification of surgical staplers from Class I (low risk) to Class II (moderate risk). Manufacturers of Class I devices are not required to submit a premarket approval notification before putting those devices on the market, meaning the FDA does not get a chance to review their safety and effectiveness.
This reclassification would require surgical staplers to undergo mandatory performance testing, demonstrations of usability and labeling requirements, among other control measures. It would be mandatory and a regulatory requirement.
The draft guidance is not mandatory, but instead lays out the FDA’s thinking on how surgical staplers should be labeled, what warnings should be added and what information gives doctors and patients the information necessary to make certain decisions about their risk-benefit profile.
Both are currently open to public comment, and Shuren also announced that the FDA will hold a public meeting of it’s General and Plastic Surgery Devices advisory panel on May 30, 2019 to discuss surgical stapler concerns and future regulation.
Get more articles like this sent directly to your inbox.
"*" indicates required fields





0 Comments