FDA Approval Requirements Removed For 1,000 Low-Risk Medical Devices
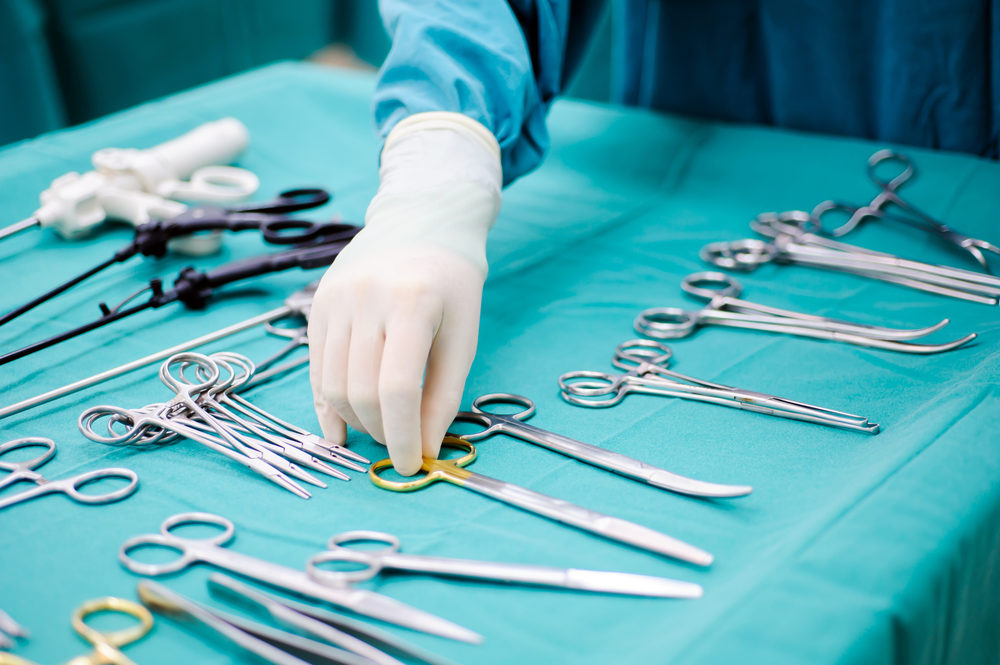
More than 1,000 medical devices considered “low risk” will no longer need to be approved by federal regulators before they are made available to doctors and patients.
The U.S. Food and Drug Administration (FDA) published a notice to the Federal Register on Tuesday, announcing a list of class II medical devices that are now exempt from both premarket approval and the FDA’s fast-track 510(k) accelerated approval process. The change, which is aimed at everyday items like dentures, acupuncture needles and menstrual pads, comes following the passage of the 21st Century Cures Act last year.
“The Food and Drug Administration…is announcing a list of class II devices that the Agency has determined based on established factors to no longer require premarket notification to provide reasonable assurance of safety and effectiveness, subject to certain limitations,” the Federal Register notice states. “The exemptions in this notice will decrease regulatory burdens on the medical device industry and will eliminate private costs and expenditures required to comply with certain Federal regulations.”

Did You Know?
Millions of Philips CPAP Machines Recalled
Philips DreamStation, CPAP and BiPAP machines sold in recent years may pose a risk of cancer, lung damage and other injuries.
Learn MoreThe 21st Century Cures Act sought to ease the way for new medical devices and new drugs, removing regulatory and financial barriers that impede them from getting to the market quickly. The latest move was required by the new law, which ordered the FDA to issue a proposed list of devices newly exempt from pre-market approval requirements within 90 days after the law’s passage. The law also requires the FDA to issue an updated list of exemptions every five years.
The agency first published the proposed list of exemptions in March.
The move comes as the FDA appears set to relax its oversight on medical devices on all fronts, not just low-risk implements.
In April, FDA officials published an editorial in the New England Journal of Medicine justifying a possible move away from requiring medical device manufacturers to conduct extensive randomized, double-blind clinical trials, which are considered the “gold standard” for medical research.
Consumer and patient safety advocates have raised concerns about steps the FDA has taken that make it too easy for untested medical devices to reach the market in the U.S. Many devices that are approved based on claims that they are “substantially equivalent” to existing devices, are marketed as featuring superior features, new manufacturing processes, materials, and surgical techniques that have never been seen or used before. Critics warn that the “21st Century Cures Act” will make it even easier for dangerous and defective medical devices to reach the market.
Rolling back regulatory oversight may further erode the ability of the FDA to protect patients from dangerous products, according to consumer advocates. A number of examples in recent years have highlighted the risks consumers face when untested devices are widely adopted in the medical field before dangerous defects are discovered based on post-marketing adverse event reports, essentially making U.S. patients unwilling test subjects.
Get more articles like this sent directly to your inbox.
"*" indicates required fields



0 Comments