BodyGuard Infusion Set Recall Issued Over Drug Delivery Problems
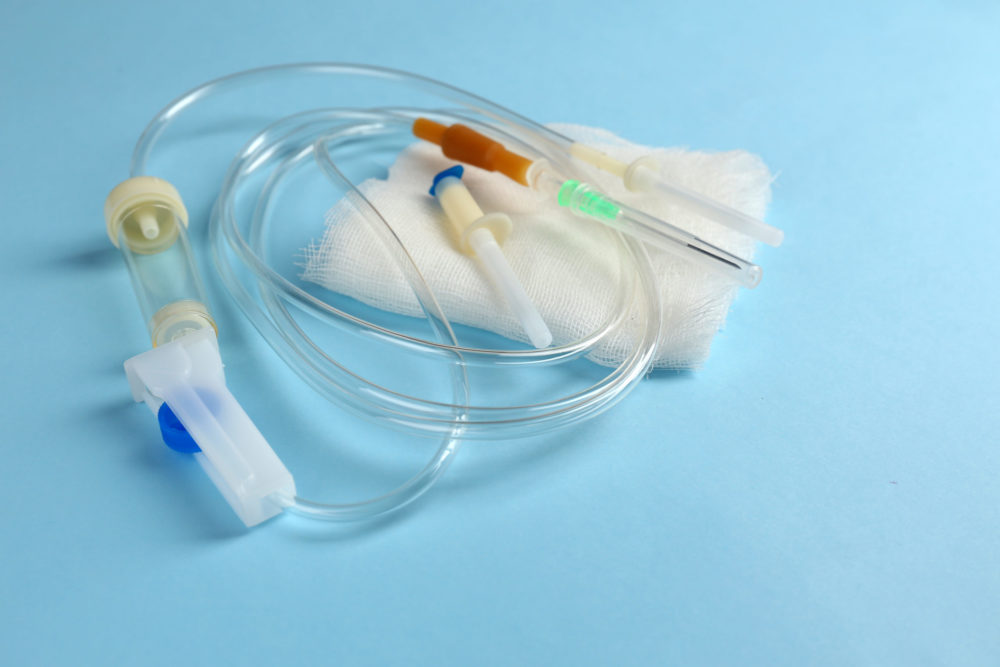
Nearly 100,000 infusion pump sets has been recalled due to a risk that defects may result in an under-delivery of drugs, posing a serious risk of injury or death for users.
The FDA announced the BodyGuard Infusion Pump System Class I recall on March 18, indicating that a manufacturing defect may cause the infusion tubing to become restricted.
Infusion pumps deliver fluids through an infusion tubing placed into a patient’s vein or through other avenues of administration. The impacted infusion pump sets are designed for use in adult, pediatric and neonatal care patients in hospitals and other healthcare facilities.

Did You Know?
Millions of Philips CPAP Machines Recalled
Philips DreamStation, CPAP and BiPAP machines sold in recent years may pose a risk of cancer, lung damage and other injuries.
Learn MoreAccording to the recall notice, the manufacturer became aware of three reports in which the Microset Infusion Sets had an extended section of tubing longer than standard lengths, which could cause the medication flow to the pumping chamber of the infusion pump to become restricted.
The FDA is warning hospitals, caregivers and patients that the occurrence of medication flow restrictions could cause the under-delivery of appropriate treatment, which could result in serious adverse health consequences including death.
A Class I recall designation means the agency believes the problems with a medical device could cause serious injuries or death. The FDA announced they are continuing to monitor adverse events reports on the BodyGuard infusion sets. To date, no injuries or deaths have been reported in relation to the recall.
The recall includes approximately 91,500 BodyGuard Infusion Pump Systems that use Microset Infusion Sets, Catalog Number A120-003XYVA. The products were manufactured by CME America, a subsidiary of Beckton Dickinson and were distributed to hospitals and healthcare facilities from October 6, 2017 through July 15, 2019.
CME America issued a letter to customers on September 16, 2019, instructing them to review their facilities inventory and discard all BodyGuard Microset Infusion Sets with catalog number A120-003XYVA.
Customers with additional questions or concerns are encouraged to contact CME America Customer and Technical Support at 303-936-4545.
The FDA is encouraging patients and healthcare personnel to report any adverse health consequences related to the recalled products to the agency’s MedWatch Adverse Event Reporting program.
Get more articles like this sent directly to your inbox.
"*" indicates required fields





0 Comments