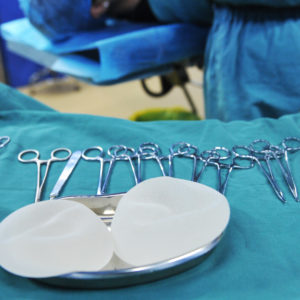
Allergan Biocell Class Action Lawsuit Claims Breast Implant Lymphoma Cases Were Not Reported To Public

Allergan continues to face a growing number of product liability lawsuits which claim its Biocell textured breast implants put women at risk of developing breast implant-associated anaplastic large cell lymphoma.
In a complaint (PDF) filed late last month in the U.S. District Court for the Central District of California, two women identified only as A.B. and C.D. seek class action status to pursue damages on behalf of themselves and all other women similarly situated with these defective and recalled implants.
The lawsuit calls on Allergan to reimburse women for the costs associated with removing Biocell breast implants, and to pay for life-long monitoring for signs of the rare cancer that may develop in the tissue surrounding the implant.
Learn More About
Women may face a risk of Anaplastic Large Cell Lymphoma (ALCL) from certain breast implants.
Learn More About this Lawsuit SEE IF YOU QUALIFY FOR COMPENSATIONAlthough neither of the plaintiffs indicate that they have developed BIA-ALCL from the Allergan Biocell implants, they indicate that they now live in perpetual fear of developing the cancer, and claim that Allergan should have to pay for all women who received the implants to have them removed and replaced and should pay for medical monitoring for the women for life.
To date, Allergan has only offered to pay for the replacement breast implants themselves, not the surgery to remove and replace its current implants, nor medical monitoring for cancer.
The Allergan Biocell class action lawsuit comes several weeks after the FDA announced that the manufacturer had agreed to issue a worldwide breast implant recall for all of products featuring the microtextured design.
The announcement came following similar recalls for Allergan breast implants in Canada, Australia and France.
The lawsuit notes that the only reason the implants are off the market is because the FDA forced the issue. Otherwise, Biocell implants would still be for sale in the U.S.
The risk of BIA-ALCL from Allergan Biocell implants was known for years, according to allegations raised in the complaint, indicating that the manufacturer chose to ignore evidence linking to textured breast implants to cancer.
“Since at least 2011, Allergan has known about the connection between its BIOCELL implants and BIA-ALCL but Allergan did not disclose the connection to consumers or the medical community,” the lawsuit states. “Instead of accurately reporting adverse events individually each time an injury or malfunction occurred, however, Allergan’s practice was to ‘bury evidence of ruptures and other injuries by reporting them as routine events that did not require public disclosure’ until 2017.”
Biocell Breast Implant Concerns
The FDA indicates that there are at least 573 known cases of BIA-ALCL worldwide, including 33 deaths. Of those, 481 have been linked to Allergan breast implants. Allergan implants were also linked to 12 of the 13 deaths where the manufacturer of the breast implant was known.
In June 2017, a study published in the medical journal Plastic and Reconstructive Surgery suggested that certain textured breast implants may increase the risk of anaplastic large cell lymphoma anywhere from 10 to 14 times, when compared to smooth breast implants.
Another study, published in October 2017, warned that many breast implant cancer cases worldwide have likely not been reported, and noted that doctors and patients may not be aware of BIA-ALCL.
The FDA is not recommending women have the breast implants removed if they are showing no symptoms. Instead, the regulators recommend recipients of these implants become familiar with the symptoms of BIA-ALCL, including persistent swelling or pain near the implant, and talk to their health care provider for further information. Those with BIA-ALCL should undergo breast implant removal and removal of the surrounding scar capsule.
The agency also recommends those who receive breast implants keep a record of the device manufacturer, unique device identifier and implant model name, which may have been provided on a patient device card from the surgeon.
Those with questions regarding the safety communication can email the Division of Industry and Consumer Education (DICE) at DICE@FDA.HHS.GOV or by calling 800-638-2041 or 301-796-7100.
Cases of BIA-ALCL or other complications should be reported to the FDA MedWatch Adverse Event Reporting Program.
Get more articles like this sent directly to your inbox.
"*" indicates required fields

1 Comments


JenniferOctober 24, 2019 at 2:40 am
I have the recalled implants but moved 9 times in the 20 years since I was implanted. I have many symptoms of BII. I am planning on explanting and feel allergan should foot some of that bill.