Arrow Intra-Aortic Balloon Catheter Kit Recall Issued After Patient Death, Bleeding Injuries
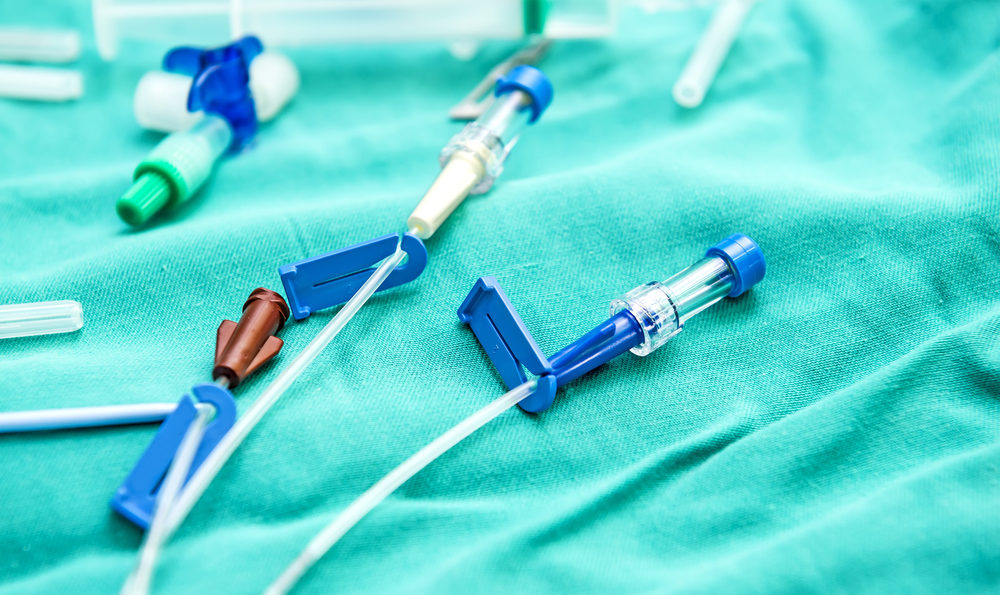
Following the death of at least one patient and severe injuries suffered by several others, federal regulators have issued a safety alert for Arrow catheter and percutaneous insertion kits used in heart patients, which are being recalled worldwide.
The Arrow Intra-Aortic Balloon (IAB) Catheter Kit recall was announced by the FDA on March 11, warning consumers and healthcare professionals about the risk of serious injuries and problems with the catheter kits and Percutaneous Insertion Kits, manufactured by Teleflex Incorporated, used on heart patients.
Reported problems with the IAB reveal the sheath body may become separated from the sheath hub. If separation occurs, a patient may bleed from the sheath. If bleeding in a patient is not promptly addressed, significant blood loss may occur, causing serious injury and death. Interruption or loss of the intra-aortic balloon pump treatment may also occur.

Did You Know?
Millions of Philips CPAP Machines Recalled
Philips DreamStation, CPAP and BiPAP machines sold in recent years may pose a risk of cancer, lung damage and other injuries.
Learn MoreTo date, at least 13 adverse events have been reported following use of the device. Six patients have been seriously injured and one patient has died resulting from use of the recalled catheter.
The catheter is inserted in the aorta and provides mechanical circulatory support for cardiac patients by inflating and deflating during different phases of the cardiac cycle. The inflating and deflating is used to increase the cardiac output and decrease the work of the heart.
The global recall affects more than 47,000 units distributed to hospitals, clinics and medical centers around the world, including the United States. Teleflex notified its domestic distributors and customers through an Urgent Medical Device recall letter on Feb. 11.
The FDA has classified this as a class I recall, which is the most serious type of medical device recall and is only issued when the product has the potential to cause severe injury or even death.
The FDA warns consumers to discontinue use of the device immediately, even if no problems have been experienced. The device should be returned to Teleflex and the FDA should be notified immediately of any problems, injuries or side effects. Consumers may contact Teleflex by calling 1-866-246-6990 or by sending an email to recalls@teleflex.com.
Want a weekly update on top lawsuits, recalls & warnings?
"*" indicates required fields

1 Comments

DonnaApril 6, 2016 at 6:08 pm
Can you inform me if the Chans/Hospice of Brunswick, Maine ever used this device?